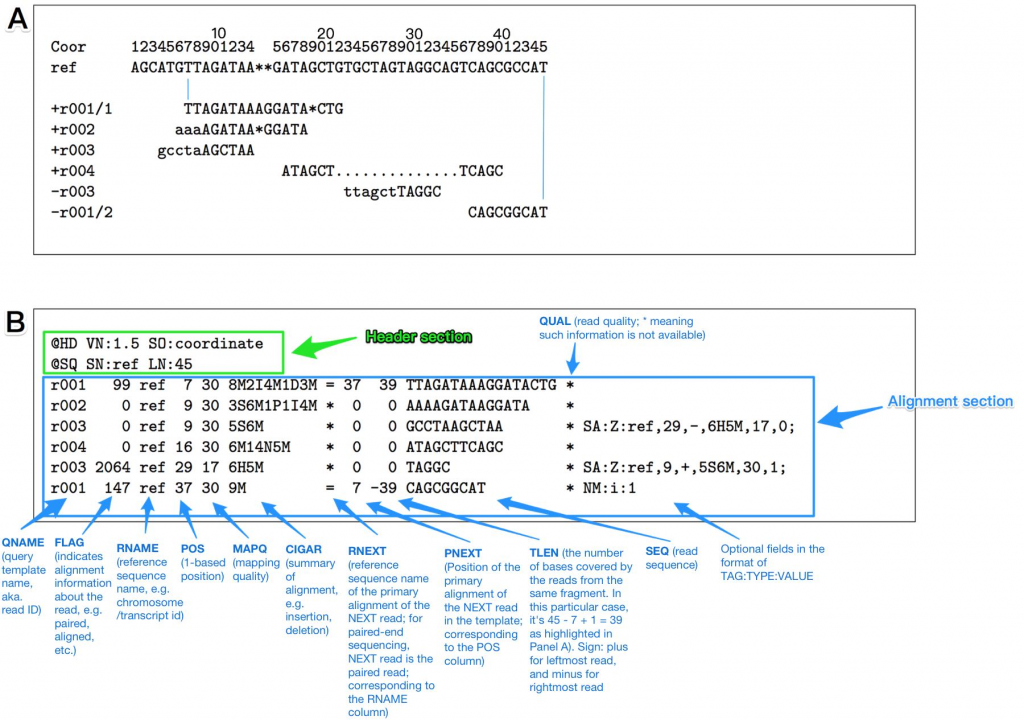
對齊記錄(Alignment Records):SAM檔的主要內容是一系列的對齊記錄,每個記錄對應一個DNA片段(讀取),描述了該片段對參考基因組的比對情況。每條對齊記錄通常包括以下信息:
未比對片段(Unmapped Fragments):如果某些DNA片段無法與參考基因組進行比對,則它們的對齊記錄可能會包含相應的信息,標記為未比對片段。
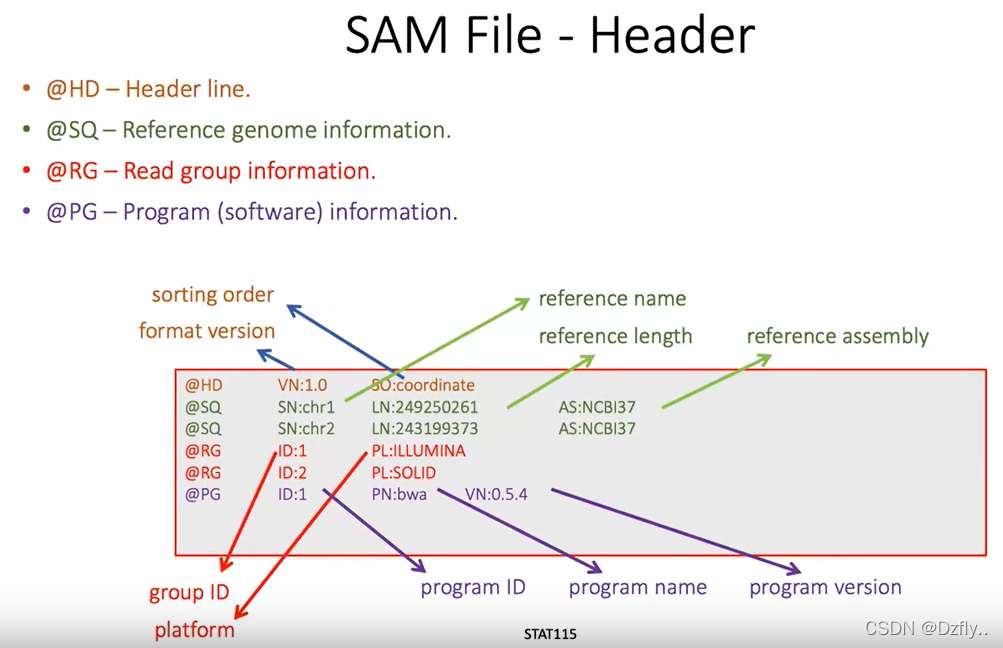
SAM檔的主要目的是存儲DNA片段對參考基因組的比對信息,並提供了關於比對質量、比對特性和其他自定義信息的詳細內容,以便進一步的分析和解釋DNA比對結果。此外,SAM格式通常與SAMtools等生物信息學工具一起使用,以進行後續的數據處理和分析。
就是 SAM 格式的壓縮二進制版本,用於存儲DNA序列比對結果的文件。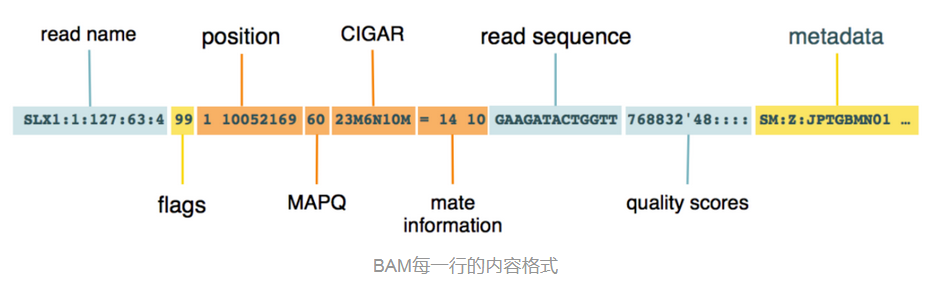
因為bam file 是二進制的檔案,所以沒辦法直接看,來一個example這是我國網(之後介紹)的一個跑完的sample,我用一個生物資訊常用的工具 samtools 來看他的檔案
samtools view -h 文件名 |less -S
samtools view -h HG002_hla_extracted.bam | less -S
![[Pasted image 20230922235726.png]]
小小題外話一下,為什麼加這段 | less -S 是因為檔案一下開啟,因為太大,回不斷往下卷,整個terminal都會被佔滿......
sam file
sam/bam格式
生信学习——sam和bam格式文件的shell小练习(附详细答案解读)


{kind=link}